- Research
- Open access
- Published:
TP53 mutations in functional corticotroph tumors are linked to invasion and worse clinical outcome
Acta Neuropathologica Communications volume 10, Article number: 139 (2022)
Abstract
Corticotroph macroadenomas are rare but difficult to manage intracranial neoplasms. Mutations in the two Cushing’s disease mutational hotspots USP8 and USP48 are less frequent in corticotroph macroadenomas and invasive tumors. There is evidence that TP53 mutations are not as rare as previously thought in these tumors. The aim of this study was to determine the prevalence of TP53 mutations in corticotroph tumors, with emphasis on macroadenomas, and their possible association with clinical and tumor characteristics. To this end, the entire TP53 coding region was sequenced in 86 functional corticotroph tumors (61 USP8 wild type; 66 macroadenomas) and the clinical characteristics of patients with TP53 mutant tumors were compared with TP53/USP8 wild type and USP8 mutant tumors. We found pathogenic TP53 variants in 9 corticotroph tumors (all macroadenomas and USP8 wild type). TP53 mutant tumors represented 14% of all functional corticotroph macroadenomas and 24% of all invasive tumors, were significantly larger and invasive, and had higher Ki67 indices and Knosp grades compared to wild type tumors. Patients with TP53 mutant tumors had undergone more therapeutic interventions, including radiation and bilateral adrenalectomy. In conclusion, pathogenic TP53 variants are more frequent than expected, representing a relevant amount of functional corticotroph macroadenomas and invasive tumors. TP53 mutations associated with more aggressive tumor features and difficult to manage disease.
Introduction
Pituitary neuroendocrine tumors are the second most common intracranial neoplasm [1]. They are usually benign, but when aggressive they may be particularly difficult to manage, accompanied by high comorbidity and increased mortality [2]. Corticotroph tumors constitute 6–10% of all pituitary tumors, but they represent up to 45% of aggressive pituitary tumors and pituitary carcinomas [2]. Functional corticotroph tumors cause Cushing’s disease (CD), a debilitating condition accompanied by increased morbidity and mortality due to glucocorticoid excess [3]. Pituitary surgery is the first line treatment, but recurrence is observed in 15–20% of cases of whom most are macroadenomas (with a size of ≥ 10 mm) [4]. Treatment options include repeated pituitary surgery, radiation therapy, medical treatment and bilateral adrenalectomy (BADX) [3]. With respect to the latter, corticotroph tumor progression after bilateral adrenalectomy/Nelson’s syndrome (CTP-BADX/NS) is a frequent severe complication and may present with aggressive tumor behavior [5,6,7].
Corticotroph tumors (including CTP-BADX/NS) carry recurrent somatic mutations in the USP8 gene in ~ 40–60% of cases [8,9,10,11,12,13]. These USP8 mutant tumors are usually found in female patients and are generally less invasive [8,9,10,11]. Additional genetic studies identified a second mutational hotspot in the USP48 gene, but no other driver mutations [14,15,16,17,18]. Focusing on USP8 wild type corticotroph tumors, we recently discovered TP53 mutations in 6 out of 18 cases (33%) [17]. Subsequent reports documented TP53 mutations in small series of mainly aggressive corticotroph tumors and carcinomas [19, 20].
TP53 is the most commonly mutated gene in malignant neoplasms [21, 22], including brain and neuroendocrine tumors [23, 24]. Until our previous report [17], TP53 mutations were only described in isolated cases of aggressive pituitary tumors and carcinomas, and were therefore considered very rare events [8, 16, 25,26,27,28]. A link between TP53 mutations and an aggressive corticotroph tumor phenotype has been hypothesized, but the heterogeneity and small size of the studies reported did not support significant clinical associations [17, 19].
To address this, we determined the prevalence of TP53 variants in a cohort of 86 patients with functional corticotroph tumors, including 61 with USP8 wild type tumors, and studied the associations between TP53 mutational status and clinical features.
Methods
Patients and samples
We analyzed tumor samples of 86 adult patients: 61 USP8 wild type and 25 USP8 mutant. Sixty-six patients (46 females, 20 males) were diagnosed with CD between 1994 and 2020 in Germany (Hamburg, Munich, Erlangen, and Tübingen) and Luxembourg. Twenty additional patients (16 females, 4 males) were diagnosed with CTP-BADX/NS, operated and followed up in 7 different international centers (Nijmegen, Munich, Erlangen, Hamburg, Paris, Rio de Janeiro, and Würzburg). Twenty-three out of 86 samples were collected prospectively between 2018 and 2021, and 63 were retrospective cases (of which 42 were investigated in the context of USP8 and USP48 screenings and published elsewhere) [9, 12, 13, 17]. Seventy-one tumors were fresh frozen and 15 were formalin fixed paraffin embedded. Paired blood was available for 12 cases. The median follow-up time after initial diagnosis was 44 months (range 2–384 months).
Endogenous Cushing’s syndrome was diagnosed according to typical clinical signs and symptoms and established biochemical procedures suggesting glucocorticoid excess. Clinical features included central obesity, moon face, buffalo hump, muscle weakness, easy bruising, striae, acne, low-impact bone fractures, mood changes, irregular menstruation, infertility and impotency. Biochemical diagnosis was based on increased 24 h urinary free cortisol (UFC) and late-night salivary cortisol levels, and lack of serum cortisol suppression after low-dose dexamethasone test. A pituitary ACTH source was confirmed by > 2.2 pmol/l (10 pg/ml) basal plasma ACTH, > 50% suppression of serum cortisol during an 8 mg dexamethasone test, and ACTH and cortisol response to corticotrophin releasing hormone stimulation.
The clinical and pathological features of our study cohort are summarized in Additional file 1: Supplementary Table 1. All patients underwent pituitary surgery. The presence of an ACTH-producing pituitary tumor was confirmed histologically after surgical resection. Biochemical remission after surgery was defined as postoperative 24 h-UFC levels below or within the normal range, or serum cortisol levels < 5 µg/dl after low-dose (1 or 2 mg) dexamethasone suppression test. Tumor control was achieved when there was no evidence of regrowth or disease recurrence. Tumor invasion was defined as radiological or intraoperative evidence of tumor within the sphenoid and/or cavernous sinuses [29]. CTP-BADX/NS was defined as an expanding pituitary tumor after bilateral adrenalectomy (BADX) following expert consensus recommendations [5].
DNA extraction, TP53 amplification and sequencing
Genomic DNA was extracted using the Maxwell Tissue DNA Kit (Promega), Maxwell Blood DNA kit (Promega) or the FFPE DNA mini kit (Qiagen), depending on the type of sample, as described previously [9, 12]. The entire coding sequence of TP53 (including exons 9β and 9γ) as well as noncoding regions adjacent to each exon were amplified using the GoTaq DNA polymerase (Promega) and specific primers (Additional file 1: Supplementary Table 2). Amplification of USP8 hotspot region and Sanger sequencing were performed as described previously [9, 12]. Chromatograms were analyzed using the Mutation Surveyor v4.0.9 (Soft Genetics). Samples were examined for TP53 coding and splicing variants. Variant position and pathogenicity was investigated in ENSEMBL (www.ensembl.org), the UCSC Genome Browser (http://genome-euro.ucsc.edu), the IARC TP53 database (https://p53.iarc.fr/TP53GeneVariations.aspx), the Catalogue Of Somatic Mutations in Cancer (COSMIC; https://cancer.sanger.ac.uk/cosmic), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), PHANTM (http://mutantp53.broadinstitute.org/), the Human Splicing Finder (HSF; http://www.umd.be/HSF3/) and VarSEAK splicing predictor (https://varseak.bio/). Variant frequencies on the general population were obtained from the Allele Frequency Aggregator (ALFA) project [30], the Genome Aggregation Database (gnomAD) [31] and the International Genome Sample Resource 1000Genome project [32]. Throughout the text, variants refer to NC_000017.11 (genomic DNA), ENST00000269305.9 (coding DNA) and ENSP00000269305.4 (protein), following the Human Genome Variation Society (HGVS) standard nomenclature system.
Statistical analysis
Statistical analysis was performed with the software package SPSS v24 (IBM). We used t-test or one-way ANOVA to analyze the association of TP53 variants with age, body mass index; Mann–Whitney U and Kruskal–Wallis to test non-parametric variables, such as tumor size, hormone levels, Ki67 index and p53 score. We corrected the analysis for multiple comparisons with the Bonferroni test. Categorical variables were analyzed using a chi-square test or Fisher exact test when needed. Survival analysis was performed using Kaplan–Meier curves with log-rank tests, and multivariate Cox regression. An exact, two-tailed significance level of P < 0.05 was considered to be statistically significant.
Results
Analysis of TP53 nucleotide variants
We analyzed all TP53 coding exons (including exons 9β and 9γ) and adjacent intronic noncoding sequences in 61 USP8 wild type tumors (49 CD and 12 CTP-BADX/NS). Of these, 13 were microadenomas (< 10 mm) and 48 macroadenomas (≥ 10 mm) at the time of the current operation. A separate group of 25 USP8 mutant tumors (17 CD and 8 CTP-BADX/NS) that were mainly macroadenomas (n = 19) was used for multiple comparison.
We found 59 variants in our cohort: 30 exclusively in USP8 wild type, 21 in USP8 mutant, and 8 in wild type and mutant tumors regardless of USP8 mutational status. No indels in the coding region of TP53 were detected. In addition, we did not find any genetic variant affecting TP53 splicing.
Nine out of 30 variants found in USP8 wild type tumors were either reported in the COSMIC database as pathogenic or absent from the common variant databases (1000Genomes, gnomAD, ALPHA) or had allele frequency < 0.0001. They were all described in cancer series: 5 as pathogenic or likely pathogenic in ClinVar, 2 as variants of uncertain significance (VUS) and 2 were not described in ClinVar (Table 1). All variants are reported to alter protein function and show clear loss of transactivation activity in a yeast based assay (Table 1) [33].
Seven variants target amino acids within the DNA-binding domain, essential for p53 activity, disrupting S2’ and S7 β-sheets or the L3 loop spatial conformation. The other two [c.1009C > G (p.Arg337Gly) and c.1031 T > C (p.Leu344Pro)] locate in the tetramerization domain and keep p53 protein as monomer impairing its transactivation activity [34]. From the 9 variants, 8 affect highly conserved p53 residues, while in c.1031 T > C (p.Met133Lys) the methionine alternates with leucine or valine among species. This variant alters protein folding, probably reducing DNA affinity [35], while the substitution of a methionine that acts as an alternative start codon abolishes the transcription of isoforms ∆133p53α, ∆133p53β and ∆133p53γ. The 9 variants were detected in nine cases (henceforth referred to as TP53 mutant; Table 1). Two tumors from unrelated patients (#6 and #7) carried the same variant c.818G > A (p.Arg273His), while one tumor (#4) carried two variants (c.718A > G and c.773A > C). Seven variants were found in heterozygosis, while the other two (from patients #1 and #2) in homozygosis. From these two, we only had paired blood/tumor samples from patient #1 and detected the variant only on the tumor sample, indicative of loss of heterozygosity (Additional file 1: Supplementary Fig. 1A). Similarly, we could demonstrate the somatic origin of the TP53 variants in four other patients with paired tumor/blood samples (#3, #5, #6 and #9).
The remaining 21/30 variants found in USP8 wild type and all 21 variants found in the USP8 mutant tumors were described as benign, likely benign or VUS with no evidence of affecting protein function. All tumors with these variants were considered TP53 wild type. From the 21 variants found in the USP8 wild type tumors (henceforth referred to as TP53/USP8 wild type group), 7 were non-synonymous variants, 8 synonymous variants and 6 non-coding variants without splicing effect. From the 21 variants found in the 25 USP8 mutant tumors, nine were synonymous, four non-synonymous and eight non-coding without splicing effect. In addition, eight variants were found in tumors regardless of USP8 mutational status that were not categorized as TP53 mutations. The intronic variant c.782 + 62G > A was found in heterozygosis in 6/70 samples. It was not reported in any database and is not predicted to have any splicing effect. The remaining seven are common variants classified as benign or likely benign in ClinVar and their allele frequencies were similar to those reported for the general population (ALFA, gnomAD and 1000Genome project) (Additional file 1: Supplementary Table 3).
Summarizing, all TP53 mutations were found in the USP8 wild type tumors, leading to a prevalence of 15% in this subgroup.
Clinical presentation of patients with TP53 mutant tumors
Patients with TP53 mutant tumors (n = 9) tended to be diagnosed at older age compared to TP53/USP8 wild type tumors (n = 52) (t-test P = 0.069; Table 2). This was significant after including the USP8 mutant group (n = 25) in the multiple comparison analysis (ANOVA P = 0.024, Table 2) and when TP53/USP8 wild type and USP8 mutant tumors were combined to a single group (TP53 wild type, n = 77; Additional file 1: Supplementary Table 4. We did not observe any sex specific predominance of TP53 mutations in contrast to USP8 mutants that are predominantly found in female patients. Furthermore, we did not find any statistically significant differences in ACTH and cortisol levels (Table2; Additional file 1: Supplementary Table 4).
Patients with TP53 mutant tumors underwent more surgeries and tumor resection was more frequently incomplete compared to TP53/USP8 wild type (Table 2). These patients also underwent a higher number of additional therapeutic procedures (radiation, n = 7; BADX, n = 4; temozolomide, n = 3; pasireotide, n = 2). Only one patient (#4) with TP53 mutant tumor, a 77 year-old man, had a single surgery without any other treatment, but his follow-up was short (< 6 months).
We observed TP53 mutations more frequently in CTP-BADX/NS (4/12, 33%) compared to CD (5/49, 10%), trending towards statistically significant difference (Fischer exact test P = 0.065 for TP53 mutant vs. TP53/USP8 wild type, P = 0.060 for comparison among the 3 groups; Table 2).
The TP53 mutant group associated with higher disease-specific mortality and shorter survival than USP8 mutant or TP53/USP8 wild type groups (log rank test, P = 0.023, Fig. 1). Three patients with TP53 mutant tumors (all CTP-BADX/NS) died of disease-related deaths: two from severe cerebral hemorrhage after surgery and stereotactic radiation and one from uncontrolled disease after five failed operations, radiotherapy (gamma knife, fractionated radiation) and chemotherapy (temozolomide, bevacizumab) at the ages of 75, 80 and 37, respectively. Ten-year survival was 27% for patients with TP53 mutant tumors, 100% for TP53/USP8 wild type and 86% for USP8 mutant. In our cohort, survival did not differ after adjusting for age (HR 7.7, 95%CI 0.6–107.7, P = 0.127).
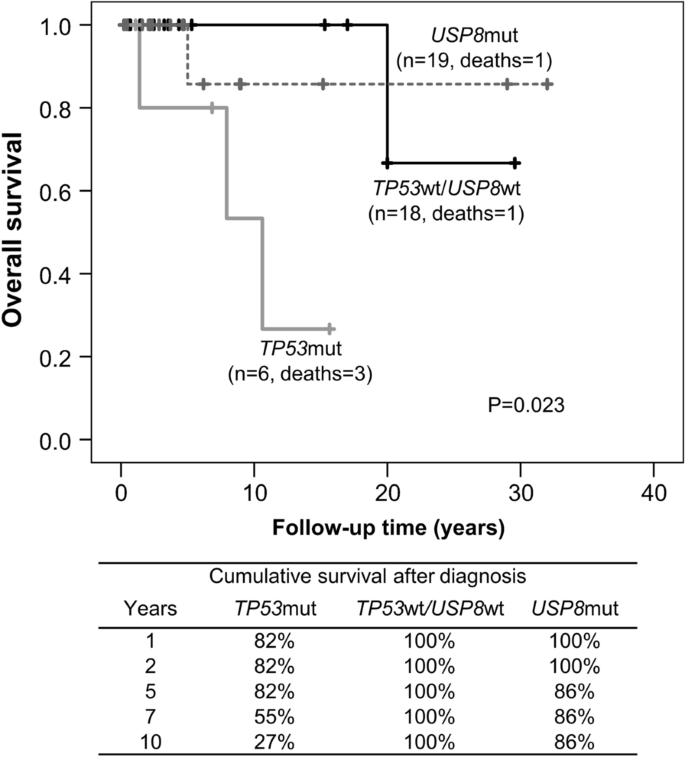
Kaplan–Meier curve showing overall survival in patients with TP53 mutant/USP8 wild type, USP8 mutant/TP53 wild type, and TP53 wild type/USP8 wild type corticotroph tumors. The table underneath the graph shows the 10-year cumulative survival after diagnosis
Tumor samples from prior surgeries were available from one TP53 mutant case (#8, Table 1). This male patient had his first pituitary surgery for CD when he was 30 years old and was treated with γ-knife one year later. He then underwent two more pituitary surgeries and BADX until the age of 35. He developed CTP-BADX/NS with para- and retrosellar tumor extension along with panhypopituitarism and underwent two more pituitary surgeries before dying at the age of 38 due to complications of the disease. We detected the TP53 variant c.1009C > G (p.Arg337Gly) in all available tumor specimens, including his first and latest surgeries (Additional file 1: Supplementary Fig. 1B).
No statistical association was found between clinical data and any of the 8 common variants.
Characteristics of TP53 mutant corticotroph tumors
All TP53 mutations were found in macroadenomas (9/66; Table 3). TP53 mutant tumors were larger that TP53/USP8 wild type (mm median [IQR] 20.0 [14.0] vs. 15.0 [14.3]), but this did not reach statistical significance (Table 3). Multiple comparison analysis showed that the difference in tumor size is significant only comparing TP53 mutant with USP8 mutant (median [IQR] 23.3 [14.0] vs. 14 [7.3] mm; Kruskal–Wallis P = 0.019; Bonferroni corrected P = 0.018).
Parasellar invasion was reported in 34 out of 64 cases, for which this information was available, and it was more common in TP53 mutant tumors (100% vs. 53% and 55% for TP53/USP8 wild type and USP8 mutant, respectively; Fischer exact test P = 0.006). TP53 mutant tumors had higher Knosp grade (Kruskal–Wallis P = 0.011) with the majority being Knosp 4 (Table 3, Additional file 1: Supplementary Table 4).
Ki67 proliferation index was available for 36 cases (6 TP53 mutant). Five out of six TP53 mutant tumors had Ki67 ≥ 3% and the overall Ki67 was higher than in the wild type tumors (Kruskal–Wallis P = 0.01; Bonferroni corrected P = 0.008 for TP53/USP8 wild type) (Table 3). Ki67 ≥ 10% was reported in 6 tumors, from which 5 were TP53 mutant (Fischer exact test P < 0.0001; the remaining case was TP53/USP8 wild type).
We had information on p53 immunostaining from 9 cases (all macroadenomas), four of which TP53 mutant: 3 tumors (from patients #5, 6 and 9) showed high p53 immunoreactivity, while the one (from patient #3) carrying a nonsense variant leading to a truncated protein was p53 negative. The five TP53 wild type cases showed isolated nuclear staining in < 1–3% of cells.
Summarizing, TP53 mutations were significantly associated with features related to a more aggressive tumor behavior, such as incomplete tumor resection, more frequent parasellar invasion, higher Knosp grade, and higher Ki67 proliferation index (Table 3; Additional file 1: Supplementary Table 4).
Discussion
Herein, we investigated the prevalence of TP53 mutations by screening a large cohort of 61 functional corticotroph tumors with USP8 wild type status, and found variants altering protein function in 15% of cases. We did not detect TP53 mutations in a separate group of 25 USP8 mutant tumors, which is in concordance with previously published small next-generation sequencing series [8, 18, 19].
Since we focused on USP8 wild type tumors, macroadenomas were overrepresented in our cohort. Consequently, it should be noted that the prevalence of TP53 mutations is expected to be lower in the general CD population. In fact, ~ 50% of corticotroph tumors carry USP8 mutations, which others and we have shown to be mutually exclusive. Corticotroph tumors with USP8 mutations are associated with female predominance, younger age at presentation, and less invasiveness (despite shorter time to relapse) [9, 11, 13, 18, 36]. In contrast, TP53 mutant tumors were diagnosed mostly at older age, did not show sex predominance and were larger and more invasive, with lower complete resection rate. None of the 19 microadenomas included in our study carried TP53 mutations. Still, we need to acknowledge that since no sample was microdissected we may have lost microadenoma cases with TP53 mutations. Instead, we found TP53 mutations in 9/66 macroadenomas (14%) and 8/34 (24%) invasive tumors, supporting the findings from smaller series [17, 19].
Tumor size at presentation or invasiveness do not reliably predict aggressiveness. Instead, the European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumors and carcinomas proposed a definition of pituitary tumor aggressiveness based on rapid or clinically relevant tumor growth despite optimal therapeutic options, along with bone invasion [37]. A recent study in a series of 9 aggressive pituitary tumors and carcinomas carrying ATRX mutations reported a high frequency of missense TP53 variants (5/9, 55.6%), further suggesting a link between TP53 mutational status and unfavorable outcome [20]. We do not have exact information on changes of tumor growth for the majority of our cases, but the higher number of surgical and radiation interventions, the higher Knosp grades, and the increased mortality rate indicate that patients with TP53 mutant tumors obviously follow a more aggressive disease course.
Ki67 proliferation index together with p53 immunostaining and mitotic count have been suggested as histological markers of pituitary tumor aggressiveness [29, 38]. In our series, Ki67 was significantly higher in TP53 mutant tumors, reinforcing our prior observation of a higher proportion of TP53 mutant tumors in the Ki67 ≥ 3 group [17]. We had limited information on p53 immunohistochemistry, since this measure is not routinely performed in our collaborative centers. Nevertheless, in the few tumors with known p53 immunopositivity, it was higher in the TP53 mutant group, which is in concordance with a previous study reporting high p53 immunoreactivity in all TP53 mutant tumors [19].
A mutagenic action of radiation on TP53 has been hypothesized by small series on radiation-induced tumors. For instance, TP53 mutations were reported in 58% of radiation-induced sarcomas [39], while a meta-analysis reported TP53 mutations in 14/30 radiation-induced gliomas [40]. A previous study reported a case with frameshift TP53 mutation in the CTP-BADX/NS tumor, but not in the initial CD surgeries, and the mutation was therefore suspected to be induced by radiotherapy [41]. In our series, however, 4 out of 7 TP53 mutant tumors were obtained before radiation.
In their case report, Pinto et al. suggested that TP53 mutations are acquired during tumorigenesis and condition tumor evolution [41]. In contrast, Casar-Borota et al. and Uzilov et al. reported high allele fraction of TP53 mutations, indicating that they are not a late event in corticotroph tumorigenesis [19, 20]. In addition, Uzilov et al. reported TP53 mutations in all tumor specimens from their two TP53 mutant cases with multiple surgeries [19]. Similarly, in our series we had tissue from multiple pituitary surgeries from one patient and found the TP53 variant in all samples (CD and CTP-BADX/NS), including specimens obtained before radiotherapy. Taken together, these observations suggest that in most cases, TP53 mutations may appear early during tumor development.
A limitation of our study is the short follow-up of patients who were prospectively included. Moreover, material from repeated surgeries was lacking from most patients with TP53 mutant tumors, hampering the examination of tumor evolution in these patients. Similarly, we had limited access to blood samples, so we could not demonstrate the somatic origin for all variants. Nevertheless, the older age at initial diagnosis of CD in patients with TP53 mutant tumors (53 ± 19.5 years old, with the youngest patient diagnosed at the age of 30) and the absence of additional neoplasias during follow-up also support a somatic instead of a germline origin. Furthermore, conditions related to germline TP53 mutations, such as Li-Fraumeni syndrome, very rarely present with pituitary tumor [42]. To our knowledge, the only published case so far was a pediatric patient with an aggressive lactotroph tumor [43].
In addition to the TP53 mutations, we detected several common variants. Variants rs59758982 and rs1042522 have been associated with increased cancer susceptibility [44, 45]. In some cancer types, the very frequent rs1042522 c.215G > C (p.Pro72Arg) alternative variant correlated to more efficient induction of apoptosis by DNA-damaging chemotherapeutic drugs, growth suppression and higher metastatic potential [46,47,48]. In nonfunctioning pituitary tumors, alternative allele C (leading to p.Arg72) was related to early age at presentation and reduced p21 expression [49]. Very recently, an overrepresentation of the rs1042522 alternative allele C (p.Arg72) was reported in 9 out of 10 corticotroph neoplasias including 5 functional tumors (allele frequency 0.900, vs 0.714 in Latino/admixed American in gnomAD [31]) without any association with clinical features [50]. In our cohort, we did not detect different allele frequencies in any of the investigated common variants (including rs1042522) compared with public databases, nor statistical association with any clinical variable, rendering their contribution to corticotroph pathophysiology unlikely.
Conclusion
Screening a large corticotroph tumor series revealed that TP53 mutations are more frequent than previously considered. Furthermore, we show that patients with TP53 mutant tumors had higher number of surgeries, more invasive tumors, and worse disease outcome. Our study provides evidence that patients with pathogenic or function altering variants may require more intense treatment and extended follow-up, and suggests screening for TP53 variants in macroadenomas with wild type USP8 status. Further work is needed to determine the potential use of TP53 status as a predictor of disease outcome.
Availability of data and materials
The authors declare that the relevant data supporting the conclusions of this article are included within the article and its supplementary information file. Additional clinical data are available from the corresponding authors MT and LGPR upon reasonable request.
Abbreviations
- CD:
-
Cushing’s disease
- BADX:
-
Bilateral adrenalectomy
- CTP-BADX/NS:
-
Corticotroph tumor progression after bilateral adrenalectomy/Nelson’s syndrome
- ACTH:
-
Adrenocorticotropic hormone
- SD:
-
Standard deviation
- IQR:
-
Interquartile range
- HR:
-
Hazard ratio
References
Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS (2018) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol 20:iv1-86
McCormack A, Dekkers OM, Petersenn S, Popovic V, Trouillas J, Raverot G et al (2018) Treatment of aggressive pituitary tumours and carcinomas: results of a European society of endocrinology (ESE) survey 2016. Eur J Endocrinol 178:265–276
Fleseriu M, Auchus R, Bancos I, Ben-Shlomo A, Bertherat J, Biermasz NR et al (2021) Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol 9:847–875
Dimopoulou C, Schopohl J, Rachinger W, Buchfelder M, Honegger J, Reincke M et al (2013) Long-term remission and recurrence rates after first and second transsphenoidal surgery for Cushing’s disease: care reality in the Munich metropolitan region. Eur J Endocrinol 170:283–292
Reincke M, Albani A, Assie G, Bancos I, Brue T, Buchfelder M et al (2021) Corticotroph tumor progression after bilateral adrenalectomy (Nelson’s syndrome): systematic review and expert consensus recommendations. Eur J Endocrinol 184:P1-16
Fountas A, Lim ES, Drake WM, Powlson AS, Gurnell M, Martin NM et al (2020) Outcomes of patients with Nelson’s syndrome after primary treatment: a multicenter study from 13 UK pituitary centers. J Clin Endocrinol Metab 105:1527–1537
Kemink SA, Wesseling P, Pieters GF, Verhofstad AA, Hermus AR, Smals AG (1999) Progression of a Nelson’s adenoma to pituitary carcinoma; a case report and review of the literature. J Endocrinol Invest 22:70–75
Reincke M, Sbiera S, Hayakawa A, Theodoropoulou M, Osswald A, Beuschlein F et al (2015) Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat Genet 47:31–38
Pérez-Rivas LG, Theodoropoulou M, Ferraù F, Nusser C, Kawaguchi K, Stratakis CA et al (2015) The Gene of the ubiquitin-specific protease 8 is frequently mutated in adenomas causing Cushing’s disease. J Clin Endocrinol Metab 100:E997-1004
Ma Z-Y, Song Z-J, Chen J-H, Wang Y-F, Li S-Q, Zhou L-F et al (2015) Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res 25:306–317
Hayashi K, Inoshita N, Kawaguchi K, Ardisasmita AI, Suzuki H, Fukuhara N et al (2016) The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur J Endocrinol 174:213–226
Pérez-Rivas LG, Theodoropoulou M, Puar TH, Fazel J, Stieg MR, Ferraù F et al (2018) Somatic USP8 mutations are frequent events in corticotroph tumor progression causing Nelson’s tumor. Eur J Endocrinol 178:59–65
Albani A, Pérez-Rivas LG, Dimopoulou C, Zopp S, Colón-Bolea P, Roeber S et al (2018) The USP8 mutational status may predict long-term remission in patients with Cushing’s disease. Clin Endocrinol (Oxf) 89:454–458
Bi WL, Horowitz P, Greenwald NF, Abedalthagafi M, Agarwalla PK, Gibson WJ et al (2017) Landscape of genomic alterations in pituitary adenomas. Clin Cancer Res 23:1841–1851
Song Z-J, Reitman ZJ, Ma Z-Y, Chen J-H, Zhang Q-L, Shou X-F et al (2016) The genome-wide mutational landscape of pituitary adenomas. Cell Res 26:1255–1259
Chen J, Jian X, Deng S, Ma Z, Shou X, Shen Y et al (2018) Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat Commun 9:3171
Sbiera S, Perez-Rivas LG, Taranets L, Weigand I, Flitsch J, Graf E et al (2019) Driver mutations in USP8 wild-type Cushing’s disease. Neuro Oncol 21:1273–1283
Neou M, Villa C, Armignacco R, Jouinot A, Raffin-Sanson ML, Septier A et al (2020) Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell 37:123-134.e5
Uzilov AV, Taik P, Cheesman KC, Javanmard P, Ying K, Roehnelt A et al (2021) USP8 and TP53 drivers are associated with CNV in a corticotroph adenoma cohort enriched for aggressive tumors. J Clin Endocrinol Metab 106:826–842
Casar-Borota O, Boldt HB, Engström BE, Andersen MS, Baussart B, Bengtsson D et al (2021) Corticotroph aggressive pituitary tumors and carcinomas frequently harbor ATRX mutations. J Clin Endocrinol Metab 106:1183–1194
Campbell PJ, Getz G, Korbel JO, Stuart JM, Jennings JL, Stein LD et al (2020) Pan-cancer analysis of whole genomes. Nature 578:82–93
Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J et al (2016) TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat 37:865–876
Horbinski C, Ligon KL, Brastianos P, Huse JT, Venere M, Chang S et al (2019) The medical necessity of advanced molecular testing in the diagnosis and treatment of brain tumor patients. Neuro Oncol 21:1498–1508
van Riet J, van de Werken HJG, Cuppen E, Eskens FALM, Tesselaar M, van Veenendaal LM et al (2021) The genomic landscape of 85 advanced neuroendocrine neoplasms reveals subtype-heterogeneity and potential therapeutic targets. Nat Commun 12:4612
Herman V, Drazin NZ, Gonsky R, Melmed S (1993) Molecular screening of pituitary adenomas for gene mutations and rearrangements. J Clin Endocrinol Metab 77:50–55
Levy A, Hall L, Yeudall WA, Lightman SL (1994) p53 gene mutations in pituitary adenomas: rare events. Clin Endocrinol (Oxf) 41:809–814
Tanizaki Y, Jin L, Scheithauer BW, Kovacs K, Roncaroli F, Lloyd RV (2007) P53 gene mutations in pituitary carcinomas. Endocr Pathol 18:217–222
Kawashima ST, Usui T, Sano T, Iogawa H, Hagiwara H, Tamanaha T et al (2009) P53 gene mutation in an atypical corticotroph adenoma with Cushing’s disease. Clin Endocrinol (Oxf) 2009:656–657
Trouillas J, Roy P, Sturm N, Dantony E, Cortet-Rudelli C, Viennet G et al (2013) A new prognostic clinicopathological classification of pituitary adenomas: a multicentric case-control study of 410 patients with 8 years post-operative follow-up. Acta Neuropathol 126:123–135
Phan J, Jin Y, Zhang H, Qiang W, Shekhtman E, Shao D et al (2020) ALFA: allele frequency aggregator: national center for biotechnology information, U.S. National Library of Medicine. Available from www.ncbi.nlm.nih.gov/snp/docs/gsr/alfa/
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q et al (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443
Fairley S, Lowy-Gallego E, Perry E, Flicek P (2020) The International genome sample resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res 48:D941–D947
Kato S, Han S-Y, Liu W, Otsuka K, Shibata H, Kanamaru R et al (2003) Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci 100:8424–8429
Kawaguchi T, Kato S, Otsuka K, Watanabe G, Kumabe T, Tominaga T et al (2005) The relationship among p53 oligomer formation, structure and transcriptional activity using a comprehensive missense mutation library. Oncogene 24:6976–6981
Greenblatt MS, Chappuis PO, Bond JP, Hamel N, Foulkes WD (2001) TP53 mutations in breast cancer associated with BRCA1 or BRCA2 germ-line mutations: distinctive spectrum and structural distribution. Cancer Res 61:4092–4097
Sesta A, Cassarino MF, Terreni M, Ambrogio AG, Libera L, Bardelli D et al (2020) Ubiquitin-Specific Protease 8 mutant corticotrope adenomas present unique secretory and molecular features and shed light on the role of ubiquitylation on ACTH processing. Neuroendocrinology 110:119–129
Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V et al (2018) European society of endocrinology clinical practice guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol 178:G1-24
Thapar K, Scheithauer BW, Kovacs K, Pernicone PJ, Laws ER (1996) p53 expression in pituitary adenomas and carcinomas: correlation with invasiveness and tumor growth fractions. Neurosurgery 38:765–70
Gonin-Laurent N, Gibaud A, Huygue M, Lefèvre SH, Le Bras M, Chauveinc L et al (2006) Specific TP53 mutation pattern in radiation-induced sarcomas. Carcinogenesis 27:1266–1272
Whitehouse JP, Howlett M, Federico A, Kool M, Endersby R, Gottardo NG (2021) Defining the molecular features of radiation-induced glioma: a systematic review and meta-analysis. Neuro-Oncol Adv 3:1–16
Pinto EM, Siqueira SACC, Cukier P, Fragoso MCBVCBV, Lin CJ, De Mendonca BB et al (2011) Possible role of a radiation-induced p53 mutation in a Nelson’s syndrome patient with a fatal outcome. Pituitary 14:400–404
Orr BA, Clay MR, Pinto EM, Kesserwan C (2020) An update on the central nervous system manifestations of Li–Fraumeni syndrome. Acta Neuropathol 139:669–87
Birk H, Kandregula S, Cuevas-Ocampo A, Wang CJ, Kosty J, Notarianni C (2022) Pediatric pituitary adenoma and medulloblastoma in the setting of p53 mutation: case report and review of the literature. Childs Nerv Syst. https://doi.org/10.1007/s00381-022-05478-8
Granja F, Morari J, Morari EC, Correa LAC, Assumpção LVM, Ward LS (2004) Proline homozygosity in codon 72 of p53 is a factor of susceptibility for thyroid cancer. Cancer Lett 210:151–157
Sagne C, Marcel V, Amadou A, Hainaut P, Olivier M, Hall J (2013) A meta-analysis of cancer risk associated with the TP53 intron 3 duplication polymorphism (rs17878362): geographic and tumor-specific effects. Cell Death Dis 4:e492
Katkoori VR, Jia X, Shanmugam C, Wan W, Meleth S, Bumpers H et al (2009) Prognostic significance of p53 Codon 72 polymorphism differs with race in colorectal adenocarcinoma. Clin Cancer Res 15:2406–2416
Dumont P, Leu JIJ, Della Pietra AC, George DL, Murphy M (2003) The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 33:357–365
Basu S, Gnanapradeepan K, Barnoud T, Kung CP, Tavecchio M, Scott J et al (2018) Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1α. Genes Dev 32:230–243
Yagnik G, Jahangiri A, Chen R, Wagner JR, Aghi MK (2017) Role of a p53 polymorphism in the development of nonfunctional pituitary adenomas. Mol Cell Endocrinol 446:81–90
Andonegui-Elguera S, Silva-Román G, Peña-Martínez E, Taniguchi-Ponciano K, Vela-Patiño S, Remba-Shapiro I et al (2022) The genomic landscape of corticotroph tumors: from silent adenomas to ACTH-secreting carcinomas. Int J Mol Sci. 23:4861
Funding
Open Access funding enabled and organized by Projekt DEAL. The study was supported by the Deutsche Forschungsgemeinschaft (DFG) (Project number: 314061271-TRR 205 to MF, MR and MT; FA 466/5-1 to MF; DE 2657/1-1 to TD), Metiphys program of the LMU Medical Faculty (to AA), Else Kröner-Fresenius Stiftung (Project number: 2012_A103 and 2015_A228 to MR) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ; Project number: E-26/211.294/2021 to MRG).
Author information
Authors and Affiliations
Contributions
LPGR and MT designed the study. LPGR, JS, AA and ST implemented the study. LGPR did the data analysis. SR, GA, TD, MF, MRG, ARH, GKS, MAT, RR, JF, MB, INK, JH, JT, WS, JH and MR provided patient materials and data. LGPR and MT interpreted the data and composed the main draft of the manuscript. All authors have seen, corrected and approved the final draft.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki and was approved by the ethics committee of the LMU Munich (Nr. 643-16). All patients provided written informed consent.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary Table 1: Description of study cohort. Supplementary Table 2: Primers used for TP53 amplification and Sanger sequencing. Supplementary Table 3: Common TP53 variants in the study cohort. Supplementary Table 4: Comparison of TP53 mutant versus TP53 wild type group. Supplementary Figure 1. Chromatograms showing the TP53 variants found in the corticotroph tumor of patient #1 and #8 (Table 1). A. The variant c.398T>A was present in homozygocity in the tumor and absent in the blood. B. The variant c.1009C>G is detected in all available surgical specimens in this patient. First and 2nd surgeries were Cushing’s disease tumors and 4th and 5th CTP-BADX/NS.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Perez-Rivas, L.G., Simon, J., Albani, A. et al. TP53 mutations in functional corticotroph tumors are linked to invasion and worse clinical outcome. acta neuropathol commun 10, 139 (2022). https://doi.org/10.1186/s40478-022-01437-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-022-01437-1